Wegener’s granulomatosis is a rare autoimmune, multisystemic disease affecting the small vessels. It is characterized by granulomatous vasculitis of the upper and lower respiratory tracts together with glomerulonephritis.

The disease is named after Friedrich Wegener, a German pathologist, who first described the full clinical picture of the disease. In 2006, two investigators from UK and US investigated Wegener’s past and discovered that he was a follower of the Nazi Regime.
Furthermore, He used to conduct experiments on Jewish people. In addition, he was wanted by the Polish authorities and his files were forwarded to the United Nations War Crimes Commission.
Because of his past, the investigators suggested that the eponym “Wegener’s granulomatosis” be abandoned. In 2011, the American College of Rheumatology (ACR), the American Society of Nephrology (ASN) and the European League Against Rheumatism (EULAR) resolved to change the name to granulomatosis with polyangiitis. However, the name “Wegener’s Granulomatosis” is still widely used and liked by medical professionals worldwide.
Wegener’s granulomatosis can be seen at any age
About 15% of patients with Wegener’s granulomatosis are less than 19 years of age, but only rarely does the disease occur before adolescence. The mean age of onset is 40 years. The disease is rarely seen in Japan and African Americans.
Pathophysiology of the Wegener’s Granulomatosis
The histopathologic hallmarks of the disease are:
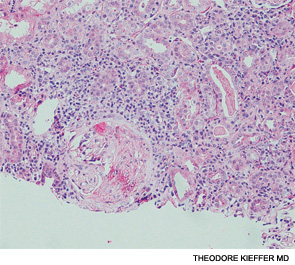
- Necrotizing vasculitis of small arteries and veins
- Granuloma formation,
The Respiratory tract and kidney involvement are common features. Lungs may have multiple, bilateral, nodular cavitary infiltrates. The upper airways may reveal inflammation, necrosis, and granuloma formation, with or without vasculitis.

Focal segmental glomerulosclerosis may be seen on Renal Biopsy in the early stages of the disease. While patients with advanced disease may manifest as Crescentic Glomerulonephritis. Characteristically, Immune complex deposition is not found.
In addition to the classic triad of the disease, virtually any organ can be involved with vasculitis, granuloma, or both.
Clinical manifestations of Wegener’s Granulomatosis:
Upper Airways Involvement:

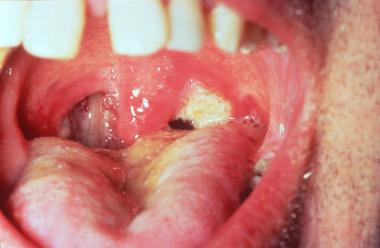
95% of the patients with Wegener’s Granulomatosis have upper airways involvement. Patients may have paranasal sinus pain and drainage. The nasal discharge may be purulent or bloody, with or without nasal mucosal ulceration.
Nasal septal perforation leading to a saddle nose deformity may be seen. Patients may have recurrent serous otitis media as a result of eustachian tube blockage. Subglottic tracheal stenosis may occur and lead to severe airway obstruction.
Pulmonary involvement in Wegener’s granulomatosis
Lung involvement occurs in 85 to 90% of the patients with Wegener’s granulomatosis. Patients may present with a cough, hemoptysis (blood in sputum), shortness of breath, and chest discomfort. Patients may also present with a lung collapse and airway obstruction.

Lung nodules and cavitations may be seen on chest x-rays or HRCT (high resolution computed tomography) of the chest.
Eye involvement:
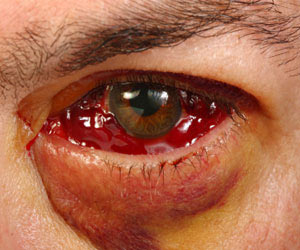
Eye involvement occurs in half of the patients with Wegener’s granulomatosis. The following eye changes may occur in these patients:
- Mild conjunctivitis to dacryocystitis,
- Episcleritis,
- Scleritis,
- Granulomatous sclero-uveitis,
- Ciliary vessel vasculitis, and
- Retroorbital mass lesions leading to proptosis
Miscellaneous findings:
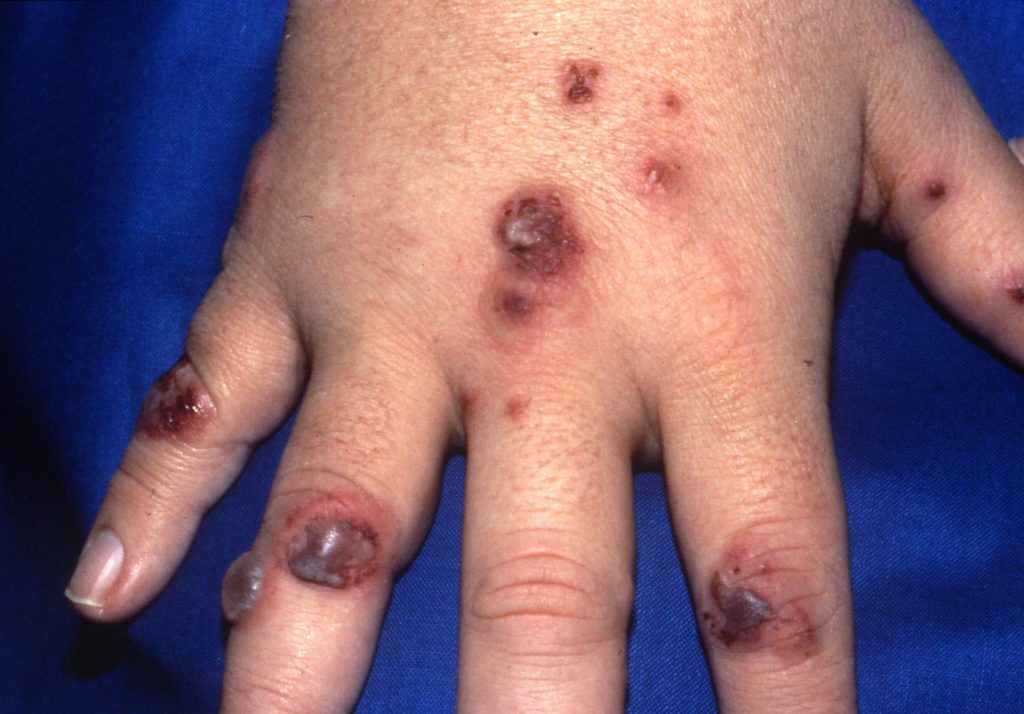
Skin lesions (develop in 46% of the patients) appear as papules, vesicles, palpable purpura, ulcers, or subcutaneous nodules; biopsy reveals vasculitis, granuloma, or both.
Cardiac involvement (occurs in 8% of the patients) manifests as pericarditis, coronary vasculitis, or, rarely cardiomyopathy.
Nervous system manifestations (found in 23% of the patients) include cranial neuritis, mono-neuritis multiplex, or, rarely, cerebral vasculitis and/or granuloma
Renal involvement in Wegener’s Granulomatosis:
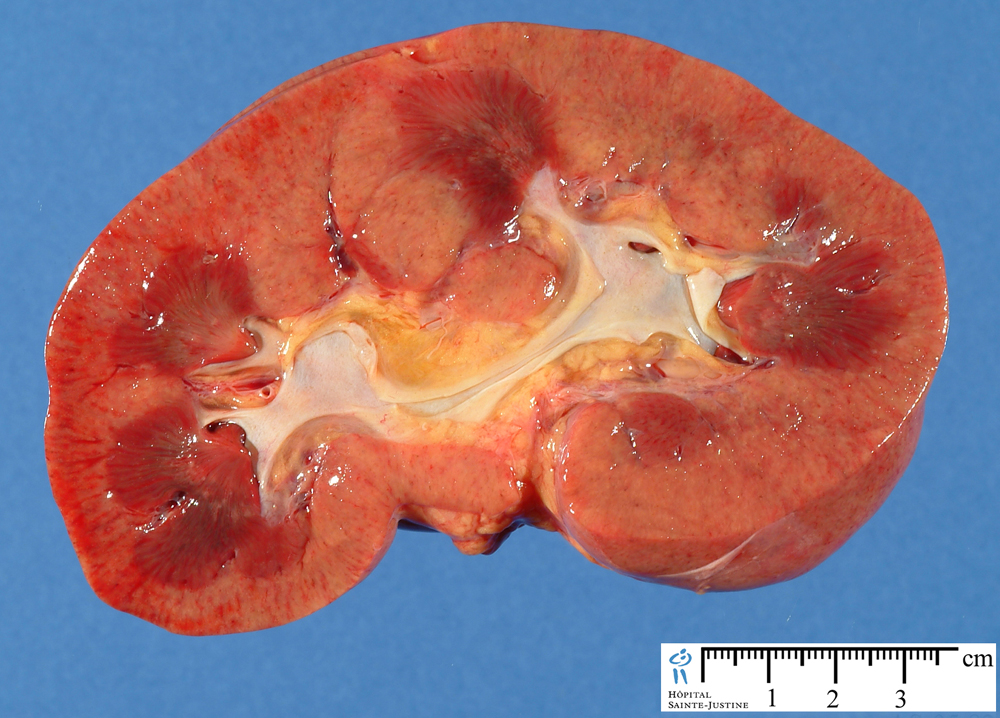
Renal involvement occurs in 77% of the patients. It accounts directly or indirectly for most of the mortality rates in this disease.
Patients can have a mild glomerulitis with proteinuria, hematuria, and red blood cell casts. Occasionally a rapidly progressive renal failure ensues unless appropriate treatment is instituted.
While the disease is active, most patients have nonspecific symptoms and signs such as malaise, weakness, arthralgias, anorexia, and weight loss.
Fever may indicate activity of the underlying disease but more often reflects secondary infection, usually of the upper airway.
Laboratory findings:

- Markedly elevated erythrocyte sedimentation rate (ESR),
- Mild anemia and leukocytosis,
- Mild hypergammaglobulinemia (particularly of the IgA class), and mildly elevated rheumatoid factor.
- Thrombocytosis may be seen as an acute phase reactant.
- 90% have a positive antiproteinase-3 ANCA if active disease (the sensitivity drops to ~60–70% if the disease is inactive)
- Increased incidence of venous thrombotic events
Diagnosis of Wegener’s Granulomatosis:

Necrotizing granulomatous vasculitis on tissue biopsy in a patient with compatible clinical features is suggestive of Wegener’s granulomatosis.
Pulmonary tissue offers the highest diagnostic yield, almost invariably revealing granulomatous vasculitis.
Biopsy of upper airway tissue usually reveals granulomatous inflammation with necrosis but may not show vasculitis.
Renal biopsy can confirm the presence of pauci-immune glomerulonephritis.
The specificity of a positive antiproteinase-3 ANCA for granulomatosis with polyangiitis (Wegener’s) is very high, especially if active glomerulonephritis is present.
False-positive ANCA titers have been reported in certain infectious and neoplastic diseases.
Treatment of Wegener’s Granulomatosis:
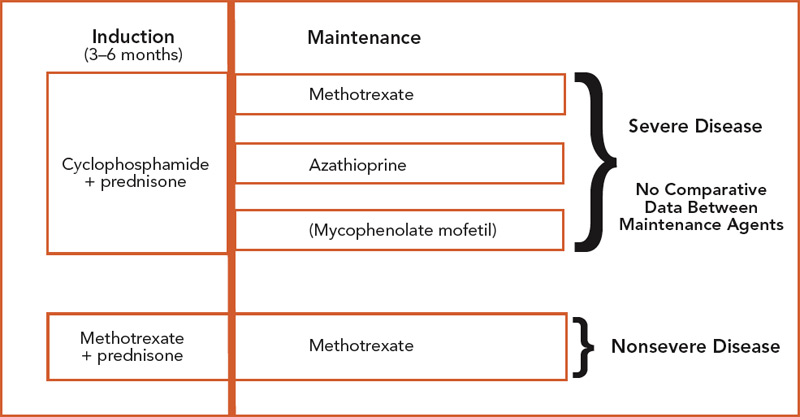
Treatment of Wegener’s granulomatosis require a multidisciplinary approach
Role of Glucocorticoids:
Glucocorticoids provide symptomatic improvement, with little effect on the ultimate course of the disease
Role of Cyclophosphamide:
Cyclophosphamide results in a marked improvement in more than 90% of patients, complete remission in 75% of patients, and 5-year patient survival in over 80%.
Other immunomodulators and immunosuppressant drugs:
Azathioprine, methotrexate and biologic agents may be used for maintenance therapy and in patients with mild disease.
Plasma exchange may be considered in patients with rapidly progressive renal disease (serum creatinine level greater than 5.65mg/dL) in order to preserve renal function.
Complications of Wegener’s Granulomatosis:
Patients can develop a varying degree of renal insufficiency, hearing loss, and chronic pulmonary dysfunction (Respiratory problems may result from upper-airway obstruction (eg, subglottic stenosis) or pulmonary involvement (eg, pleural effusion, dyspnea, diffuse alveolar hemorrhage [DAH])
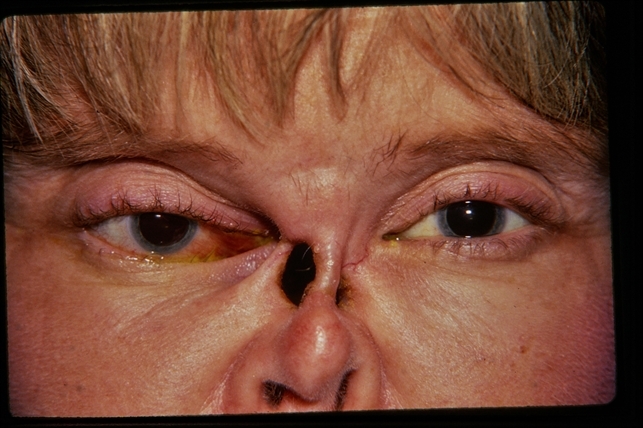
Patients may develop a saddle nose deformity due to cartilage destruction and can have a chronically impaired sinus function.
Myocardial infarction has been reported with Wegener’s granulomatosis due to coronary vasculitis.
Treatment-associated complications:
These include:
- Hemorrhagic cystitis due to cyclophosphamide therapy
- Osteoporotic fractures due to steroid use
- Urothelial (bladder) cancer – associated with cyclophosphamide therapy
- Myelodysplasia, and Avascular necrosis.
The most common causes of death in GPA are as follows:
- Infection
- Respiratory and renal failure
- Malignancy
- Cardiovascular events